



 Copy to clipboard
Copy to clipboard 
Sensor-loaded devices, bacteria-clearing solutions, unique implant materials—these are technologies that take years, even decades to develop before entering and evolving the orthopedic market. To provide patients and providers with timely access to ground-breaking medical devices, FDA has made moves to speed up its development, assessment and review of novel products through the Breakthrough Devices Program.
The program is voluntary. Products need to meet specific criteria, be it breakthrough technology, no approved or cleared alternative regulatory pathways or offers significant advantages over existing products, to be approved. While the program is intense, if approved, manufacturers can enjoy VIP-like benefits in their communication with FDA as well as Medicare backing to alleviate reimbursement hurdles.
As of the end of 2020, orthopedics accounted for 6% of the devices that received the designation since the program launched in 2016, according to FDA. Unlike 510(k)s or PMAs, FDA doesn’t publicly disclose companies that receive the designation. However, we’ve tracked 10 orthopedic companies and 11 products that have been granted access to the Breakthrough program in the last 18 months.
To keep a finger on the pulse of innovative orthopedic technologies close to entering or on the market, and to understand the hurdles they’ve cleared thus far, we highlight those companies.
Tiny Sensors CHIRP in Tibial Stems
Canary Medical, CHIRP
From fridges to watches, the internet of things (IoT) has made its way into so many aspects of modern daily life. With billions of everyday items connected to the internet, why aren’t life-saving medical devices? Why should medicine be any different? Bill Hunter, M.D., asked himself those questions before he started Canary Medical in 2012.
“My Apple Watch knows where it is on the planet, keeps track of my steps and does all kinds of other things,” he said. “I’m a doctor by training, so it seemed strange to me that we put in all this medical hardware and none of it provides any feedback. My refrigerator can talk to the internet; how come my hip or knee or coronary stent can’t do the same?”
The Canary Health Implantable Reporting Processor (CHIRP) is currently implemented in the company’s lead product, the Persona IQ, a knee stem that attaches to the Persona Personalized Knee from Zimmer Biomet. While on the outside, it looks no different than a typical stem that directly inserts into the tibial extension, the implant’s dead space holds a tiny sensor, initially designed for drones.
The sensor is powered by a pacemaker battery with a low draw that enables operation for 20 years or more, matching most implants’ lifespan. Data is transferred through radio waves, a reliable system that’s been used by pacemakers for decades. Patients can see data like how many steps they’re taking, distance traveled, stride length and more through a small home base station. Meanwhile, a more sophisticated physician interface allows for viewing range of motion and other factors to determine whether or not the patient is walking normally, saving unnecessary trips to the office.
Most of Canary’s team have backgrounds in cardiology, so the technology takes inspiration from uninterrupted ambulatory cardiac monitoring that allows physicians to follow heart rhythms weeks after surgery. While the concept of having a continuous patient feedback loop has been implemented in other disciplines, Dr. Hunter said that Canary is the first to bring internally generated data into orthopedics.
Canary’s FDA reviewer suggested applying for Breakthrough Device Designation, which the company received in October 2019. Dr. Hunter said that working with FDA was helpful.
“They felt self-reporting implants was an obvious next evolution in total joints, so they had set up different offices to handle data science and algorithms,” he said. “CHIRP was really the first of its kind going through, so they wanted to be especially interactive with us, asking and answering questions in real time, because other people may be following this pathway. It’s also allowed us to break things down and learn as we go along.”
Dr. Hunter anticipates FDA clearance very soon, with a potential product launch coming in 1H21. Canary’s technology could be part of a broader wave of transforming medical devices, which is exciting, said Dr. Hunter.
“If this is something with predictive value, physicians could use the analytics to determine which patients are recovering well and which are at risk for infection or complications,” he said. “It’s going to take a few thousand patients and time to determine that, but being able to detect an aneurysm or heart valve function is also an exciting thought.”
Personalized Implants for Spine Deformity
Carlsmed, aprevo
Carlsmed was founded in 2018 with headquarters in Carlsbad, California. CEO and Co-founder Mike Cordonnier has held various leadership roles for large medtech companies and, as an engineer by trade, he understands the value of turning data into devices. He believes that using data to personalize medical devices to correct adult spinal deformities can result in better outcomes for patients. Carlsmed’s aprevo is the only FDA cleared Breakthrough Device for spine on the market today.
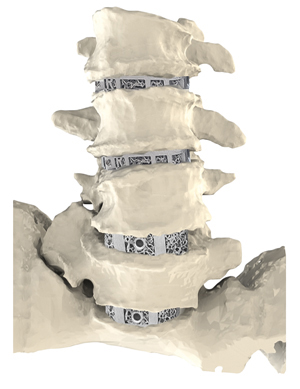
“Stock devices require the surgeon to choose from a large inventory of implants,” he said. “Our aprevo patient-specific IBF devices are personalized to match the planned correction.”
Carlsmed is mainly focused on correcting debilitating adult spine deformities, which can decrease spinal surgery complications. The proprietary aprevo technology uses patient imaging information and prior outcomes data to create Personalized Deformity Correction with Patient Specific spinal implants.
aprevo is reportedly the first implant ever to receive both Breakthrough Device Designation and 510(k) market clearance from FDA (December 2020). With the device currently on the market, Cordonnier said the opportunity to have guaranteed national Medicare coverage and the New Technology Add on Payment (NTAP) is a great advantage of taking FDA’s Breakthrough pathway.
“Having Medicare guarantee national coverage of the devices will go far in incentivizing adoption and increasing provider utilization,” he said.
Cordonnier said that it was helpful working with FDA through the submission process.
“We appreciate their help in making this new technology available to patients and providers,” he said. “We have a massive opportunity ahead of us to transform the standard of care using data to personalize surgery and devices for each patient to optimize outcomes.”
Electrical Stimulation to Eliminate Biofilm
Garwood Medical Devices, BioPrax
Total knee and hip replacements are two of the most commonly performed surgeries in the United States, with more than 1.9 million cases annually. About 2.2% of those become infected. While this may seem like a small number, the impacts on these thousands of individuals can be devastating. Up to 82% of early intervention treatments to address the infection fail, some of which result in amputation or death, according to research cited by Garwood Medical.

“The five-year mortality rate is higher than for many cancers,” Wayne Bacon, President and CEO of Garwood Medical, said. “The Gold Standard is a two-stage revision, a devastating surgery to remove the implant, debride tissue and bone, then treat with antibiotics. About half of these patients never end up with a replacement implant, and over one in five die within five years. Even worse is the five-year mortality rate for the early-intervention (Debridement Antibiotics and Implant Retention) DAIR procedure, which exceeds 50%. The cost to families and society is massive.”
Bacon founded Garwood Medical in Buffalo, New York, in 2014 and licensed the technology behind BioPrax in 2016. The electrical stimulation treatment is thought to help eliminate biofilm infections on knee implants more effectively than antibiotics alone. Two skin-based electrodes adhere like those used during an EKG, while a small needle makes contact with the implant to complete an electrical circuit. The corresponding electrochemical reaction produces hydrogen peroxide and hydroxides to drive up the pH to a bactericidal level that breaks down the biofilm and kills bacteria.
“Even if a surgeon opens up the area, they can only wash what they can see,” Bacon said. “They can’t see behind the implant at the bone or where tissue is hiding part of the implant, but BioPrax can treat 100% of the implant’s surface, which is unique.”
BioPrax is under investigation to be used alongside the DAIR standard of care after the patient is closed, “cleaning up” any remaining bacteria.
BioPrax was accepted into FDA’s Breakthrough Device Program in October 2019. FDA indicated that the technology is a good candidate for the De Novo classification as well, which would lower the Class from three to two. The designation is also welcome news for investors as the company completes its Series C financing.
Garwood Medical has started pre-clinical studies through a grant at the University of Buffalo’s Jacobs School of Medicine. It will begin its safety study in March, hopes to apply for Investigational Device Exemption in June and be first-in-human in November. This year should be busy for the company, which is also partnering with the University at Buffalo and the Walter Reed National Military Medical Center through another grant to study infection control and skin integration with transdermal implanted prosthetics.
Garwood plans to expand the BioPrax technology to all artificial joints. After gaining initial market acceptance, Bacon believes that Garwood Medical can obtain FDA clearance for one additional implant per year.
“We’ve done years of work on the technology behind BioPrax,” he said. “We’re collecting data along the way to see if we can use it for preventing infection and treating hips, shoulders and other metal devices anywhere in the body.”
Breakthrough Designation Numbers Grow in Orthopedics
The Breakthrough Designation has been awarded to more than a handful of additional orthopedic companies within the last two years. Their technologies span orthopedics, from knee to spine to orthobiologics, and also FDA submission types, from 510(k)s, De Novos and Premarket Approvals.
Active Implants, NUsurface
Founded: 2004
Headquarters: Memphis, Tennessee
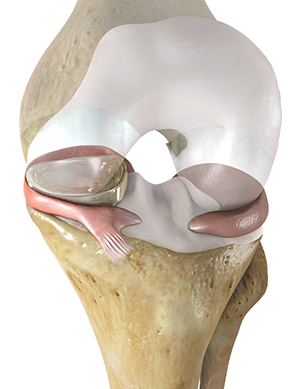
Active Implants’ NUsurface meniscus is intended to treat patients with persistent knee pain following medial meniscus surgery. As a result of its unique medical-grade polymer, composite structure and design, it does not require fixation to bone or soft tissues. The implant mimics the function of the natural meniscus and redistributes loads transmitted across the knee joint. NUsurface is available in Belgium, Germany, Italy and Israel and received FDA Breakthrough Designation in September of 2019. The company seeks clearance in early 2021 as it works with FDA through the De Novo review process.
AgNovos Healthcare, AGN1 LOEP SV Kit
Founded: 2012
Headquarters: Rockville, Maryland

AgNovos Healthcare’s AGN1 local osteo-enhancement procedure small volume (LOEP SV) contains a calcium-based resorbable tri-phasic implant material formulated to couple the pace of resorption to bone formation, allowing treated vertebrae immediate, substantial protection. The AGN1 LOEP SV Kit is an investigational device intended to minimally-invasively treat painful but stable vertebral compression fractures often caused by bone loss associated with osteoporosis. The kit contains the instruments and components necessary to prepare the fractured vertebral body for injection. The device is thought to give clinicians a new tool to treat stable vertebral compression fractures in a more biologically-congruent fashion. A kit is already approved in Europe for larger volume applications at other anatomical sites. AgNovos received FDA Breakthrough designation for the kit in March 2020.
CartiHeal, Agili-C
Founded: 2009
Headquarters: Israel and New Jersey

CartiHeal’s Agili-C implant is intended to treat cartilage lesions in both arthritic and non-arthritic joints. The implant is CE Marked for use in cartilage and osteochondral defects, and has been implanted in more than 500 patients with knee, ankle and toe cartilage lesions in a series of clinical trials in Europe and Israel. It was granted FDA Breakthrough designation in October 2020 and is the subject of an Investigational Device Exemption clinical study to evaluate the Agili-C implant compared with microfracture and debridement, the current standard of care. Final study results are expected in 2021.
Geistlich Pharm, Chondro-Gide
Founded: 1851
Headquarters: Wolhusen, Switzerland
Chondro-Gide collagen membrane is used in a single-step cartilage repair technique. The highly purified collagen membrane leverages the body’s own healing potential. It is used with bone marrow stimulation to treat defects. More than 10 years of clinical success in Europe have shown the product to be a cost-effective single surgery treatment for repairing lesions with bone marrow stimulation techniques.
Locate Bio, CognitOss and Chondro3 Graft
Founded: 2001
Headquarters: Nottingham, England

Locate Bio’s CognitOss presents an alternative to surgical debridement, long-term, high-dose administration of antibiotics and bone grafting, which often requires a second surgical procedure to remove the non-resorbable graft. CognitOss uses the same bone graft substitute as Locate Bio’s CertOss, a new class of composite collagen. FDA Breakthrough was granted in January 2021, while first-in-human studies and FDA 510(k) submission are slated for 2022.
Chondro3 is currently in development as a biomimetic graft for osteochondral lesions. The three-layered, proprietary collagen-based biodegradable matrix can be delivered in a single procedure, in an outpatient setting and at an affordable price. Chondro3 is designed to provide a scaffold for cellular and tissue in-growth and osteochondral defect repair at the site of lesion, supporting biomimetic repair of both cartilage and bone. The designation was granted in February 2021.
NovoPedics, MeniscoFix
Founded: 2013
Headquarters: Princeton, New Jersey
NovoPedics’ MeniscoFix is a polymer fiber-reinforced scaffold for total meniscus replacement designed to be gradually resorbed by the body and replaced by new tissue. Its fiber-reinforced design is similar to the native meniscus. The device is attached to both soft tissue and bone. In contrast to partial meniscus replacement devices, MeniscoFix is uniquely designed to be securely fixed within bone tunnels and to support mechanical loads along the entire implant, making it potentially useful even in patients with a compromised meniscal rim. It was granted Breakthrough designation in September 2020. NovoPedics received a $4.42 million grant from the U.S. Department of Defense to support further pre-clinical development at Rutgers University.
Orteq Sports Medicine, ACTIfit
Founded: 2003
Headquarters: London, England

Orteq’s ACTIfit offers a unique alternative to partial meniscectomy. The biodegradable scaffold is the result of polymer science and tissue regeneration technology developed in the Netherlands. ACTIfit’s polymer is a biodegradable polyurethane and is processed by solvent casting/porogen leaching to create a porous scaffold. ACTIfit is approved under the European CE Mark and in Korea and was granted FDA Breakthrough designation in April of 2020. Orteq is taking steps toward FDA 510(k) clearance. Though it is uncertain how soon the ACTIfit could be cleared for commercialization in the U.S., Orteq hopes that its extensive body of scientific and clinical evidence from Europe will support a timely approval.
FDA received 807 requests for the Breakthrough designation, as of December 31, 2020. Of those requests, 6% were for orthopedic products. We expect more orthopedic companies with innovative technologies to pursue this pathway.
Kathie Taylor is an ORTHOWORLD Contributor.



